




.jpg)
.png)










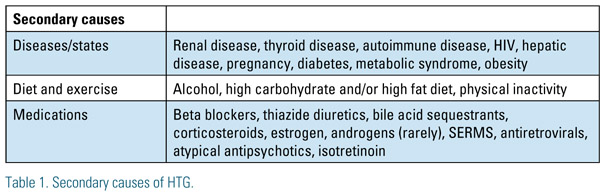
Hypertriglyceridemia (HTG) is defined as an excess of triglycerides in the blood. Primary HTG is caused by one or more genetic defects leading to triglyceride elevation. Secondary HTG is acquired; the causes are plethoric and can be identified by methodically assessing the factors listed in Table 1. Primary HTG should be suspected if a secondary cause cannot be identified. In addition, an astute clinician should consider primary HTG when the patient with a secondary cause has a fasting triglyceride measurement greater than 300 mg/dL, since primary HTG may not reveal itself without a secondary "insult" added to the clinical milieu.
 The National Cholesterol Education Program (NCEP) Adult Treatment Panel III (ATP III) guidelines recommend first treating low density lipoprotein cholesterol (LDL-C) to goal and then targeting nonhigh density lipoprotein cholesterol (non- HDL-C), unless triglycerides are ≥ 500 mg/dL. At a level of ≥ 500 mg/dL, the patient is believed to be at risk for acute pancreatitis and lowering triglycerides is the initial goal.¹ Data on triglyceride levels and their independent correlation with cardiovascular disease is unclear; however, some types of primary HTG convey higher risk for cardiovascular disease based on the specific lipoprotein elevation. Understanding the types of primary HTG, as well as distinctive responses to differences in treatment, allows the clinician to better assess and manage patient risk for pancreatitis and cardiovascular disease.
The National Cholesterol Education Program (NCEP) Adult Treatment Panel III (ATP III) guidelines recommend first treating low density lipoprotein cholesterol (LDL-C) to goal and then targeting nonhigh density lipoprotein cholesterol (non- HDL-C), unless triglycerides are ≥ 500 mg/dL. At a level of ≥ 500 mg/dL, the patient is believed to be at risk for acute pancreatitis and lowering triglycerides is the initial goal.¹ Data on triglyceride levels and their independent correlation with cardiovascular disease is unclear; however, some types of primary HTG convey higher risk for cardiovascular disease based on the specific lipoprotein elevation. Understanding the types of primary HTG, as well as distinctive responses to differences in treatment, allows the clinician to better assess and manage patient risk for pancreatitis and cardiovascular disease.
Evaluation
It can be challenging to definitively identify the genetic basis of a patient’s primary HTG as genetic testing is not readily available and the diagnostic waters are muddied when a secondary component is involved. However, serum lipid levels, personal and family history, and physical exam may guide the clinician to a likely genetic defect. First, consider the patient’s serum levels. Does the patient have an elevation in triglycerides and total cholesterol or solely triglycerides? Second, does the patient or a family member have a history of pancreatitis or cardiovascular disease? Third, are there any findings on physical exam suggestive of a specific type of primary HTG?
If a Patient Has Isolated HTG Consider:
Familial hyperchylomicronemia (Fredrickson Type 1)—Patients with Familial hyperchylomicronemia have triglycerides > 1000 mg/dL, sometimes exceeding 10,000 mg/dL, but often have normal total cholesterol. The ratio of triglyceride to cholesterol is often cited as being 10:1.2 The severe elevation in fasting chylomicrons results from a genetic mutation causing an absence of Lipoprotein Lipase (LPL), which metabolizes chylomicrons and very low density lipoprotein (VLDL), or a genetic defect in a co factor, protein or enzyme that obliterates LPL activity.3 Pancreatitis is common. Patients with Familial hyperchylomicronemia have low risk for coronary artery disease (CAD) since the elevated lipoproteins, chylomicrons, carry small amounts of cholesterol. They can exhibit eruptive xanthomas, hepatosplenomegaly and lipemia retinalis. This is a rare disorder and symptoms usually manifest during childhood.
Familial hypertriglyceridemia (Fredrickson Type 4)—Patients have normal to slightly elevated total cholesterol with triglycerides ranging from 200-1000 mg/dL. VLDL is elevated, which may result from a LPL gene mutation that decreases LPL activity. Apo B is normal.2 A secondary component, such as alcohol or high carbohydrate diet, is a common finding. Patients are usually asymptomatic and have low risk for atherogenicity.4
If a Patient Has HTG and Elevated Total Cholesterol Consider:
Familial combined hypercholesterolemia (Fredrickson Type 2B)—Patients with Familial combined hypercholesterolemia usually have triglycerides between 150-500 mg/dL and total cholesterol between 200 and 400 mg/dL. A secondary component such as insulin resistance or metabolic syndrome is common. The disorder is thought to be polygenic and large phenotypic variation can exist between family members.5 Apo B is usually elevated (> 120mg/dL), which distinguishes it from Familial hypertriglyceridemia.2 Patients have low risk for pancreatitis, but are at risk for CAD due to LDL and VLDL elevations. They do not have xanthomas.
Dysbetalipoproteinemia (Fredrickson Type 3)—Triglycerides and total cholesterol levels are elevated and equal in a one to one ratio, resulting from the Apolipoprotein (Apo) E isoform Apo E2/E2. Since many people with isoform Apo E2/ E2 do not demonstrate dyslipidemia, it is thought that a secondary cause contributes to this phenotype. An elevation in VLDL remnants increases CAD risk.4 It is thought that targeting non-HDL-C, instead of Apo B, may have more clinical utility.2 Orange palmar xanthomas, elbow and knee tubero-eruptive xanthomas and peripheral vascular disease may be found on exam.
Primary mixed hyperlipidemia (Fredrickson Type 5)—Like Familial hyperchylomicronemia, patients with Primary mixed hyperlipidemia have triglyceride levels >1000 mg/dL. Unlike Familial hyperchylomicronemia, the patient’s total cholesterol levels are also elevated and the disorder usually manifests in adulthood. Chylomicrons and VLDL elevations result from a partial deficiency of LPL or Apo C- II. They are at risk for CAD and pancreatitis. They can exhibit eruptive xanthomas, hepatosplenomegaly and lipemia retinalis.6
The appearance of a patient’s plasma can also aid in diagnosis. If a patient with severe HTG has not responded to medications, plasma should be visually assessed after phlebotomy. If plasma is clear, as opposed to cloudy or lactescent, glycerol kinase deficiency should be considered.7 Glycerol kinase deficiency, an X linked genetic disorder, causes an accumulation of free glycerol in plasma. During triglyceride synthesis, three fatty acids attach to one glycerol molecule. Most labs count glycerol instead of triglycerides when calculating triglyceride levels, thus overestimating triglycerides if free glycerol is elevated. If one suspects glycerol kinase deficiency, the clinician should seek a lab that controls for glycerol concentrations.8
Treatment
Secondary Causes
Treating secondary causes of HTG will improve triglyceride levels, thus requiring less medication. Pregnant women with triglycerides > 350 mg/dL should be referred to a high risk obstetrician and lipid specialist since pancreatitis can increase fetal and maternal morbidity and mortality.9 Examining the patient’s prescription list may reveal medicines that increase triglycerides, some of which may be substituted for a lipid neutral medication. For example, the older beta blockers may be replaced by newer beta blockers, carvedilol or nebivolol.
Diet and Exercise
Alcohol abstinence is recommended for the long-term prevention of pancreatitis.10 Fat restriction is extremely important for patients with Familial hyperchylomicronemia, since they may not respond to medications. When hyperchylomicronemia is severe, fat should be restricted to < 15% of total calories per day.¹ Medium-chain triglycerides, available as cooking oil, can provide fat that does not allow chylomicron formation since it bypasses the intestine.11 Since Familial hypertriglyceridemia, Dysbetalipoproteinemia and Familial combined hyperlipidemia can be exacerbated by metabolic disease or poor diet, it is important to counsel patients about alcohol, fat and carbohydrate intake. 2,12,13 Replacing trans-fatty acids with monounsaturated or polyunsaturated fat lowered triglyceride levels in a meta-analysis of controlled dietary trials.14 A low carbohydrate diet (<50% of calories from carbohydrates) should be recommended.1,2 Dietary fiber and complex carbohydrates should be emphasized and simple carbohydrates should be avoided.1,2 Weight loss, if needed, should be encouraged as a weight loss of 5-10% may result in a 20-30% reduction in triglycerides.15
Exercise increases lipoprotein lipase and decreases hepatic lipase activity, both which lower triglycerides. 16 Unfortunately, patients with primary HTG may not experience the same decrease in triglycerides as someone with secondary HTG. However, an exercise program should still be recommended based on patient age and abilities.
 Pharmacologic Treatment
Pharmacologic Treatment
When a patient’s triglyceride level is < 500 mg/dL, the clinician will first target LDL-C and non-HDL-C. The medications used to lower LDL-C and non-HDL-C also lower triglycerides to varying degrees. Once the patient has met his or her LDL-C and non-HDL-C goals, or if a patient presents with triglycerides > 500 mg/dL, fibric acids, omega-3 fatty acids and niacin, used primarily for triglyceride lowering, should be considered. Fibric acids are often prescribed initially, although omega-3 fatty acids may be favored in women of childbearing age. Doses at or above 4 grams daily of docosahexaenoic acid and eicosapentaenoic acid (DHA/ EPA) are usually necessary. If triglycerides are above 400 mg/dL, omega-3 fatty acids and fibric acids can increase LDLC, although shifting the pattern to larger less dense low density lipoprotein (LDL) particles and still lowering non-HDL-C. Niacin lowers LDL-C and increases high density lipoprotein cholesterol (HDL-C), in addition to lowering triglycerides and non-HDL-C, but should be used with caution in patients who have pre-diabetes or metabolic syndrome since it can worsen glycemic status.
Orlistat is a gastric and pancreatic lipase inhibitor which is FDA approved for weight loss and maintenance. Dietary fat cannot be hydrolyzed and undigested triglycerides are not absorbed.17 Orlistat has successfully lowered triglycerides in patients with Familial hyperchylomicronemia when used as an adjunct to dietary therapy.18 It has been suggested that the triglyceride-lowering effect of omega-three fatty acids or medium chain triglycerides may be lost if administered simultaneously.18,19
When managing a patient with elevated cholesterol and HTG, it is important to remember that bile acid sequestrants can increase triglycerides and are contraindicated at triglyceride level > 500 mg/dL. Their effect on triglyceride level should be monitored for those with triglycerides > 300 mg/dL.
Does the Diagnosis Matter?
If patients with primary HTG are treated to goal, some would argue that the specific genetic defect causing the HTG is not significant. However, the lipoprotein elevation that results from the genetic defect is significant when cardiovascular disease and pancreatitis risk is considered. If the clinician believes that their patient has an elevation in the atherogenic lipoproteins, chylomicron remnants, VLDL, VLDL remnants and LDL, then the clinician may focus on cardiovascular disease prevention and screening.20 If the clinician suspects an elevation in chylomicrons, then pancreatitis is a great concern. In addition, nuances in dietary and pharmacological treatment affect response between types of primary HTG. Lastly, a discussion with patients about the likelihood of a genetic basis for their disease may compel family members to be tested and treated.
Disclosure statement: Ms. Milne has received honoraria related to speaking from Kowa Pharmaceuticals America. She has received consulting fees from Amarin Corporation, Aegerion, and Genzyme.
Article By:

Cardiac Vascular Nurse and Family Nurse Practitioner
Bellevue Hospital Lipid Clinic
New York, NY
Diplomate, Accreditation Council for Clinical Lipidology
Comments
Treatment
Challenging to treat sometimes! I wonder what the next best treatment is after icosapent ethyl, fenofibrate, and statin therapy in the patient's who aren't good candidates for niacin and what triglyceride level we should be reasonably be targeting.