





.jpg)
.png)










Abstract
This case illustrates a common presentation of a rare phenomenon that should be highlighted so that clinicians are aware of the signs, implications, and management of LpX syndrome. Patient XY is a 30-year-old nonsmoker who was referred to our lipid clinic because of newly recognized severe hypercholesterolemia. She was diagnosed with acute lymphoblastic leukemia at age 28, went through several cycles of chemotherapy, and ultimately required allogeneic stem cell transplant. Her course was complicated by steroid-refractory biopsy-proven graft versus host disease (GVHD) of the liver. A lipid profile was obtained as part of routine survivorship maintenance of care and revealed total cholesterol (TC) 555 mg/dL, high-density lipoprotein cholesterol (HDL-C) 147 mg/dL, triglyceride (TG) 131 mg/dL and low-density lipoprotein cholesterol (LDL-C) 382 mg/dL (calculated by Friedewald formula). She was diagnosed with LpX syndrome as a consequence of GVHD-induced cholestatic disease. Her primary oncology team is monitoring and continues to treat the GVHD.
LpX is characterized by severe hypercholesterolemia in an individual with suspected or known obstructive liver (biliary) disease. LpX results from impaired synthesis and secretion of bile salts, and impaired negative feedback on cholesterol synthesis and LDL particle hepatic uptake. There is subsequent formation and secretion of an apolipoprotein B (apoB)-depleted hepatic micelle, rich in phospholipids and free cholesterol. With LpX, TC levels can rise to greater than 1000 mg/dL, can be associated with tendinous xanthomae, xanthelasmae, and potential arterial plaque formation. Hyperviscosity syndromes (rare manifestation of a rare disease) can also occur and may represent an urgent indication for treatment. Typical cholesterol-lowering pharmacotherapy tends to be less effective and relatively contraindicated due to the concomitant liver disease, but can be used with close monitoring if needed. Selective LDL-apheresis may not be as effective or as well tolerated as the preferred plasma exchange if immediate treatment is needed. Ultimately, treatment of the underlying liver disorder is required to effectively reestablish premorbid cholesterol levels.
Introduction
Individuals who present with severe hypercholesterolemia (low density lipoprotein cholesterol [LDL-C] levels greater than 190 mg/dL) require evaluation for etiology because of the prognostic implications associated with various causes. Severe hypercholesterolemia can be seen in a variety of common scenarios including primary monogenic disorders (familial hypercholesterolemia and sitosterolemia), polygenic conditions (common hypercholesterolemia), epigenetic, acquired conditions (thyroid dysfunction, nephrotic syndrome, advanced liver disease, extreme weight gain/loss), medication side effects, and as a consequence of dietary practices (overconsumption of saturated fats, obesity, or the flip-side, anorexia nervosa and ketogenic diets).1 However, the severest manifestations with total cholesterol (TC) levels greater than 400 mg/dL with normal triglycerides (TG) is quite rare (1 in 300,000 to 400,000 people) and found as a consequence of very specific circumstances, including genetic/epigenetic, or acquired defects.2 Homozygous familial hypercholesterolemia (HoFH), compound or double heterozygous familial hypercholesterolemia (HeFH), sitosterolemia, and acquired causes including nephrotic syndrome or lipoprotein X (LpX) can all cause this phenotype.3
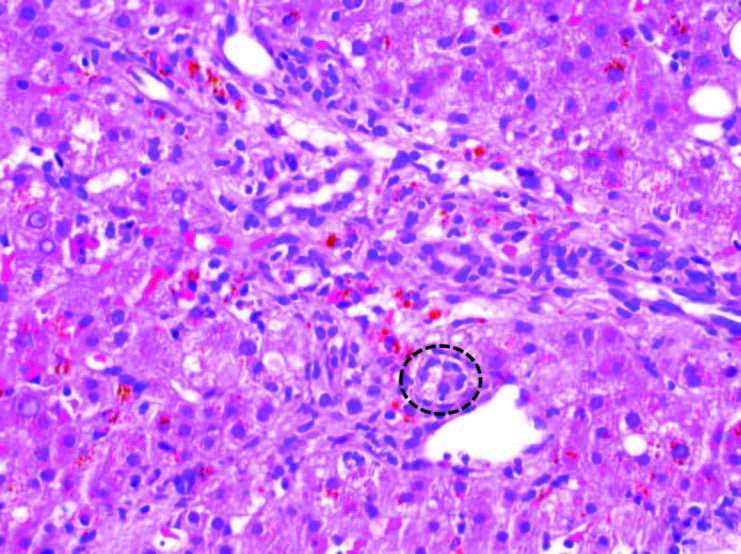
Figure 1. Liver, biopsy (200X magnification). Graft-versus-host disease. One of the injured bile ducts within this portal tract is highlighted by the black circle. The other bile ducts seen in this portal tract show similar features (see text for description).
The clinical diagnosis of a patient with severe hypercholesterolemia should be evident on the basis of historical and physical exam features, confirmed by laboratory testing, including a urine dipstick test, comprehensive metabolic panel, thyroid testing, present and historical lipid profiles, and may include genetic testing in the right context. Several different commercial laboratories offer affordable sequencing of the LDLR, APOB, PCSK9, LDLRAP genes as part of a panel which can confirm the FH genotype and apolipoprotein E (apoE) testing for the most common alleles (E2, E3, E4) is available at most commercial laboratories if needed. Specific testing for phytosterol levels with markers of absorption, and direct measurement of apolipoprotein B (apoB), TC, and TG can narrow the diagnosis further as outlined in the algorithm proposed by de Graaf et al. Free cholesterol (FC) and cholesterol ester (CE) fractionation can be done by special laboratories when LpX is expected, and there should be predominance of FC in this condition. And finally, polyanion precipitation testing can be done to confirm the presence of LpX, though a clinical diagnosis can be assumed typically after the above testing has been completed.
LpX is characterized by severe hypercholesterolemia in an individual with suspected or known obstructive liver (biliary) disease. LpX results from impaired synthesis and secretion of bile salts, and impaired negative feedback on cholesterol synthesis. There is subsequent formation and secretion of an apoB-depleted hepatic micelle, rich in phospholipids and FC.5,6 With LpX, TC levels can rise to greater than 1000 mg/dL, can be associated with tendinous xanthomae, xanthelasmae, and potential arterial plaque formation. Hyperviscosity syndromes can also occur due to excess lipoprotein and may represent an urgent indication for treatment. Typical cholesterol-lowering pharmacotherapy tends to be less effective and relatively contraindicated due to the concomitant liver disease, but can be used with close monitoring. Selective LDL-apheresis may not be as effective or as well tolerated as plasma exchange, which can be offered if urgent treatment is required.7 Therapeutic plasma exchange should probably be reserved for symptomatic hypercholesterolemia (ie, hyperviscosity syndrome [a rare manifestation of a rare condition], painful xanthomae, neuropathy). Ultimately, treatment of the underlying liver disorder is required to effectively reestablish premorbid cholesterol levels.
Case Report
Patient XY is a 30-year-old nonsmoker who was referred to our lipid clinic because of newly recognized severe hypercholesterolemia. She was diagnosed with acute lymphoblastic leukemia in May of 2015 when she was 28-years-old. She achieved clinical remission after two cycles of induction chemotherapy. She completed a total of four cycles of cyclophosphamide, vincristine, doxorubicin, and dexamethasone along with dasatinib and intrathecal chemotherapy and was started on maintenance therapy with daily dasatinib, prednisone and vincristine. At that time, although allogeneic stem cell transplant would have been standard of care, that approach was not available to her due to limited insurance coverage.
Unfortunately, she developed relapse of her disease in February of 2017 at which point she received salvage chemotherapy with blinatumomab and intrathecal chemotherapy and achieved her second clinical remission in May of 2017. In June of 2017 she was referred for a matched unrelated donor allogenic stem cell transplant with cyclophosphamide and total body radiation condition with post-transplant cyclophosphamide for graft versus host disease (GVHD) prophylaxis. Her course was complicated by steroid-refractory biopsy proven GVHD of the liver requiring experimental treatment with ruxolitinib and ursodeoxycholic acid for symptom relief. The liver biopsy revealed bile duct injury, the histologic hallmark of hepatic GVHD. The small bile ducts within the portal tracts show a disorganized arrangement of epithelial cells around an irregular lumen, cytoplasmic vacuolization, and occasional infiltration of lymphocytes (Figure 1). Fortunately, by the time we saw her, the primary oncology team already expressed that the GVHD was improving and expected resolution with treatment.
Despite oncologic stabilization, a lipid profile in September of 2017 (done as part of routine survivorship maintenance of care) revealed TC 555 mg/dL, HDL-C 147 mg/dL, TG 131 mg/dL and LDL-C 382 mg/dL (calculated by Friedewald formula). She was started on simvastatin 20 mg daily and referred to our lipid clinic for further evaluation and treatment.
There was no previous history of hypercholesterolemia and no family history of this. There was normal kidney function and urinalysis and she had a normal thyroid stimulating hormone level. Her weight had been stable and there were no extraordinary diet changes. She had normal vital signs, jaundice was noted, and there were no xanthomae or xanthelasmae and the remainder of her physical examination was normal. An apoB level and lipid profile were completed at her visit. Her lab results are shown in Table 1. Measurement of free versus esterified cholesterol content in the plasma is diagnostic wherein the fraction of cholesterol ester compared to total cholesterol is less than the normal 60-80%. This test was not performed in this case due to her improving clinical status and the strength of the clinical diagnosis and limitations of her insurance reimbursement for special testing.7
She was diagnosed with LpX syndrome caused by GVHD-induced cholestatic disease. Her primary oncology team is monitoring and continues to treat the GVHD. The simvastatin was discontinued and her clinical condition and lipid profile will be monitored.
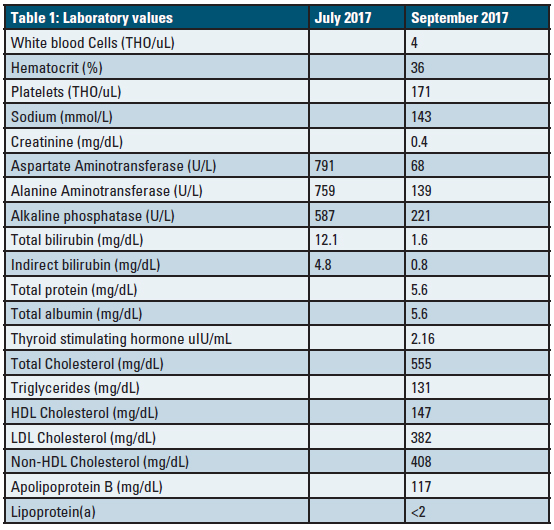
Table 1. Laboratory values from the time of visit at the lipid clinic. Liver function test values are provided at an additional time point (July 2017) at their peak for comparison.
Discussion
We describe LpX syndrome in a patient with cholestatic GVHD after allogeneic hematopoietic stem cell transplantation. LpX syndrome is a rare disorder of secondary severe hypercholesterolemia that was first described in the 1950s associated with cholestatic liver disease. It is characterized by elevated TC and LDL-C levels with relatively normal TG, and uncommonly, xanthomae, retinal cholesterol thromboembolism, corneal deposits, cholesteroloma of the lung, pseudo-hyponatremia, neuropathy, and hyperviscosity syndrome.8 Although first identified in patients with extrahepatic biliary obstruction from gallbladder disease, LpX syndrome has since been reported in the setting of intrahepatic biliary obstruction (primary biliary cirrhosis/cholangitis and primary sclerosing cholangitis), intravenous administration of intralipid emulsion for total parenteral nutrition, bone marrow transplant as in this case presentation, infants with premature liver function, and in hereditary lecithin-cholesterol acyltransferase deficiency.5,8-10
LpX is composed of phospholipid (66%), free unesterified cholesterol (22%), albumin and apolipoprotein C (6%), and the remainder is TG and cholesterol esters (6%). Unlike LDL, which is a product of catabolism of very low-density lipoprotein (VLDL), LpX is directly synthesized by hepatocytes. LpX does not contain apolipoprotein B (the primary ligand for hepatic LDL-receptors), and clearance is mediated by a non-ligand-receptor splenic mechanism.2,5,6 Confirmatory diagnostic testing involves measurement of cholesterol esters in the serum. A diagnostic test exists at the Mayo Clinic, which measures the percentage of cholesterol esters compared to total cholesterol in serum.11 The absence of esterified cholesterol in the serum (<60-80%) is diagnostic of LpX syndrome. Due to the patient’s lack of severe symptoms and improving clinical course, and health insurance limitations this specialized test was not performed in this case.
Reports of LpX syndrome are described in patients with cholestasis. In these patients, LpX was postulated by Manzato and colleagues to form as a consequence of bile reflux into plasma resulting in complexes of bile acid and albumin creating a lipoprotein complex similar in density to LDL-C.6 This was supported by in vitro experiments wherein human bile combined with whole serum containing albumin from healthy volunteers resulted in formation of LpX with a similar gel electrophoresis migration pattern to LpX in serum of patients with cholestatic liver disease. Formation of LpX was re-demonstrated in peripheral venous samples of blood from dogs as soon as three hours after creating artificial anastomoses from the bile duct to the inferior vena cava.6
Paradoxically, cholesterol synthesis is intensified in the setting of LpX syndrome. Several theories regarding this mechanism have been suggested, including absorption of cholesterol from hepatocytes into LpX particles and thus causing increased hepatic cholesterol synthesis possibly mediated by inappropriate up-regulation of 3-hydroxy-3-methyl-glutaryl-coenzyme A (HMGCoA) reductase.9 The current understanding of LpX formation through bile acid and albumin complexes is challenged based on these hypotheses and recent data showing negligible amounts of bile acid in LpX particles. Heimerl et al suggest that the relative absence of feedback inhibition on cholesterol synthesis of LpX upregulates cholesterol and phospholipid synthesis in hepatocytes which are then released into the circulation as LpX.12 The exact mechanism remains to be determined.
In several cases of LpX due to a cholestasis from either intra or extrahepatic biliary obstruction, resolution of cholesterol and LDL-C levels is demonstrated after treatment of the underlying disease or liver transplantation.5,8,13,14 However, while in excess, the clinical impact and atherogenicity of elevated cholesterol in the form of LpX remains somewhat unclear. The natural history of plaque formation due to high serum LDL-C is through oxidation of the LDL-C and inhibition of fibroblast growth factor 2 ultimately causing apoptosis of endothelial cells and inflammatory invasion. Chang et al examined isolated LDL-C/LpX in patients with primary biliary cholangitis and exposed it to copper, an oxidizing agent, and found that serum in patients with cholestatic disease did not become oxidized compared to normal serum. Combination of LpX serum with copper failed to reduce fibroblast growth factor 2 levels, which was used as a proxy for aortic endothelial cell survival. Addition of LpX serum to oxidized LDL-C serum blunted the reduction in fibroblast growth factor 2 and thus exerted a protective effect on the endothelial cells.15 On a larger scale, a systematic review of three prospective trials of patients with primary biliary cholangitis and LpX syndrome noted that over an average of 10 years, the number and cause of deaths were not disproportionately due to cardiac causes. However these were all relatively small studies of patients with only one type of disease, limiting the generalizability of this finding.13
Management of LpX syndrome involves treatment of the underlying disease if possible; for example, transplant is needed for intrahepatic cholestasis causes or treatment of rejection in cases of GVHD.5,8,16 Pharmacologic reduction in LpX with statins has little utility, but has been attempted with some success. Considering the paradoxical increase in cholesterol synthesis, statins could potentially reduce LpX formation, but their primary mechanism for LDL-C lowering (up-regulation of LDL-R) may not be as important in this condition since these particles do not seem to be cleared by LDL-R. Importantly, several statins including lovastatin, simvastatin, atorvastatin, and rosuvastatin are cleared in bile and can accumulate to toxic doses in cases of cholestasis.13,17 Ursodeoxycholic acid has been shown to reduce serum cholesterol by 16-19% without any change in HDL-C or TG levels.13 While this is a modest reduction, the symptomatic benefits in minimizing pruritis related to cholestasis may be beneficial. There are three case reports of plasma exchange resulting in successful treatment of LpX levels and normalization of lipid panels in patients with primary sclerosing cholangitis and one with GVHD-induced LpX syndrome. Because this is a rare condition, case reports and case series represent the only clinical therapeutic evidence.8,16-18
In this case presentation, our patient has steroid-refractory GVHD now improving with experimental treatment with ruxolitinib. We elected to discontinue simvastatin due to potential for adverse effects and modest, if any, benefit given her youth and relatively low risk from atherosclerotic cardiovascular disease. We will closely monitor her clinical status and lipid panel as the GVHD improves. If needed, we can offer her treatment with plasma exchange, but this will likely be reserved for the development of more severe symptoms (eg, hyperviscosity syndrome).
This case illustrates a common presentation of a relatively rare cause of a rare condition and opportunity to review the clinical signs, implications, and management of LpX syndrome.
Disclosure statement: Dr. Gopal has no disclosures to report. Dr. Fortuna has no disclosures to report.
Dr. Soffer has received honoraria from Aegerion, Potomac CME, ACP Pier, and MD Consult. He has served as a local subinvestigator.
References available here.
Article By:

Cardiology Fellow Physician
University of Pennsylvania
Philadelphia, PA

Internal Medicine/Clinical Lipidology
University of Pennsylvania Health System
Philadelphia, PA
Diplomate, American Board of Clinical Lipidology

Hepatic Pathology Fellow
University of Pennsylvania
Philadelphia, PA