




.jpg)
.png)










An exciting new approach to treating obesity and insulin resistance involves significant carbohydrate restriction with the goal of achieving a ketogenic state that results in weight loss and improved insulin sensitivity. Many lipidologists, however, have seen frequent cases of worsening lipid and lipoprotein parameters with such carbohydrate-restricted diets. What’s going on here, what are the associated risks of this increased atherogenic lipoprotein burden for cardiovascular disease (CVD), what’s the mechanism, and what should be done with these patients? Here’s a typical case scenario:1
A healthy, 55-year-old menopausal female, who is 5’4”, 154 pounds, and has no cardiovascular disease history struggles with weight loss, decides to follow a low- carb/Paleo diet. Her baseline lipid panel is: total cholesterol = 196, low-density lipoprotein cholesterol (LDL-C) = 105, high-density lipoprotein cholesterol (HDL- C) = 75, triglycerides (TG) = 78, and non-HDL-C = 121 (all in mg/dL). She loses 30 pounds in three months on this diet and her new lipid panel is: TC = 323, LDL-C = 230, HDL-C 83, TG 49, and non-HDL-C= 240 (all in mg/dL). Total low-density lipoprotein particle number (LDL-P) (by NMR) = 2643 nmol/L (99th percentile population cut point). Is this presumed increase in atherogenic lipoproteins (no baseline lipoprotein values other than that inferred by unremarkable non-HDL-C) dangerous?
 Patients commonly go on a low-carb ketogenic diet (LCKD) (defined by many at less than 50 grams of carbohydrate or <10 percent of calories/day) to lose weight, with the secondary goal of improving insulin sensitivity and decreasing cardiometabolic risk. The Paleo diet2 is another variation of a LCKD, often but not necessarily with a major increase in saturated fat intake. It should be noted that many popular variations of LCKD exist, including Atkins, South Beach, Paleo, Dukkan, and others, and that these diets may vary substantially in fat and other macronutrient composition. There is a substantial body of evidence that a LCKD is at least as effective at weight loss as low- fat diets, and changes in the triglyceride/ HDL axis and markers of insulin resistance are usually more beneficial with low- carbohydrate diets. It is tempting to conclude that these favorable changes would be associated with a reduction in cardiovascular disease but, as of yet, we have no randomized clinical trial data with CVD outcomes to support this.3
Patients commonly go on a low-carb ketogenic diet (LCKD) (defined by many at less than 50 grams of carbohydrate or <10 percent of calories/day) to lose weight, with the secondary goal of improving insulin sensitivity and decreasing cardiometabolic risk. The Paleo diet2 is another variation of a LCKD, often but not necessarily with a major increase in saturated fat intake. It should be noted that many popular variations of LCKD exist, including Atkins, South Beach, Paleo, Dukkan, and others, and that these diets may vary substantially in fat and other macronutrient composition. There is a substantial body of evidence that a LCKD is at least as effective at weight loss as low- fat diets, and changes in the triglyceride/ HDL axis and markers of insulin resistance are usually more beneficial with low- carbohydrate diets. It is tempting to conclude that these favorable changes would be associated with a reduction in cardiovascular disease but, as of yet, we have no randomized clinical trial data with CVD outcomes to support this.3
The most consistent and predictable lipid change on LCKD is a decrease in triglycerides. Changes in other lipid parameters have been variable, but an increase in LDL-C often has been noted (anecdotally, about 30 percent of the time) in clinical practice and is thought to be related to the increased saturated fat composition of the LCKD. A 2009 meta-analysis comparing low-carbohydrate diets (defined as < 45 percent of calories from carbohydrates) vs. low-fat diets (<30 percent of calories from fat) for six months or more, showed that “compared with participants on low-fat diets, persons on low-carbohydrate diets experienced a slightly but statistically significantly lower reduction in total cholesterol and low-density lipoprotein cholesterol, but a greater increase in high-density lipoprotein cholesterol and a greater decrease in triglycerides.4” Data on lipoprotein changes with LCKD are more sparse and variable. A six-month study in children placed on a ketogenic diet for epilepsy showed a significant and persistent increase in Apolipoprotein B (ApoB).5 Other studies have shown reductions in ApoB; for example, this study of lipid and lipoprotein changes in overweight men with atherogenic dyslipidemia.6 Volek, et al., discuss improvements in the ApoB/ ApoA-1 ratio in adult subjects on a LCKD vs. a low-fat diet.7 However, it is difficult to draw conclusions from all of these studies because of the heterogeneity of the subjects and composition of the diets studied.
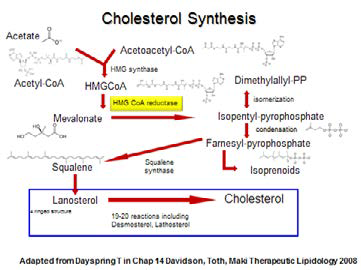
What is the etiology of the increase in LDL-C/ApoB/LDL-P in our patient? The expected drop in triglycerides in such a diet would obviously change the composition of LDL lipoproteins, causing an increase in their size and cholesterol composition and a shift from small dense “pattern B” to large and buoyant “pattern A.” The magnitude of this change is dependent on the starting triglyceride level and genetic factors. Most LCKD followers also increase their dietary saturated fat (SF) intake, which is a recognized cause for an increase in LDL-C levels. The explanation for the ApoB/LDL-P increase from carbohydrate restriction is more complex.8 When dietary carbohydrate restriction occurs, the body switches to fatty acid (FA) catabolism for energy. Intracellularly, these FA are activated to form acyl-CoA, and then acetyl-CoA, which eventually is utilized for fuel production via the Krebs cycle. Acetyl-CoA may be converted into ketone bodies, which can be measured in the urine as a simple index of the degree of ketosis the dieter has achieved on a LCKD. (For a further discussion of the safety and clinical utility of ketogenic diets, see Peter Attia’s excellent discussion,9 “Ketosis – advantaged or misunderstood state? [Part I]).” Two acetyl-CoA may combine to form acetoacetyl-CoA, which can be utilized to form mevalonic acid and, ultimately, increase cholesterol synthesis through the 3-hydroxy-3-methyl-glutaryl-CoA reductase (HMG-CoA) pathway. Also, when ketone bodies are in excess, there is an increased production of HMG-CoA, again driving the cholesterol synthetic pathway. Anecdotally, it has been observed that many on LCKD with increased LDL-C/ ApoB/ LDL-P have increased desmosterol levels, a marker for increased cholesterol synthesis. Diabetic ketoacidosis and anorexia nervosa patients have elevated LDL-C/ApoB/LDL-P, in part via similar mechanisms. Excessive hepatic cholesterol synthesis will drive the formation of more ApoB particles, and the increased synthesis in the liver will decrease the expression of the LDL-receptor (modulated through the nuclear transcription factor “sterol toxicity sensor” Liver X Receptor [LXR]), prolonging LDL clearance, and adding to the increased LDL-P concentration in circulation.
One might wonder if the accompanying increase in exogenous cholesterol intake occurring in many LCKD diets — increased eggs, shellfish, meats, etc. — might drive increases in LDL-C, but it now is well established that absorption of endogenous cholesterol (of hepatobiliary origin) plays a much greater role in determining LDL-C levels than cholesterol from exogenous sources.
So what is our patient’s risk from her increased LDL-C/ApoB/LDL-P and what should we do about it? Certainly we can speculate that, in a typical overweight and insulin-resistant patient, improvements in her insulin selectivity and “vascular biology” from the LCKD and its associated weight loss would help reduce her cardiometabolic risk. We don’t know this patient’s metabolic status, but the low-TGHDL-C ratio argues against much insulin resistance. No one knows what the CVD risk of increased LDL-C/ApoB/LDL-P is in LCKD states, and the evidence for the long-term safety of this dietary approach is lacking. Many patients with increased ApoB/LDL-P (for example, familial hypercholesterolemia patients) never get cardiovascular disease, and the explanations for this are all speculative. Sarah Hallberg, DO, of Indiana University is the principal investigator for a large study comparing patients with type 2 diabetes or prediabetes who are being treated with a ketogenic diet vs. patients being treated using the standard American Diabetes Association (ADA) dietary guidelines, looking at NMR LDL-P, metabolic markers, and carotid intima- media thickness (cIMT) over two years. She said, “Atherogenic dyslipidemia so dramatically improves and diabetes resolution occurs so frequently that we have to be asking, even in the patients who have a rise in LDL-C, are we not still improving their health? For those patients with a rise, is there a perfect blend with statin therapy?” However, it is difficult to ignore the massive epidemiologic and clinical trial data that increased ApoB/LDL-P is associated with increased CVD risk and that lowering these parameters with statin therapy reduces risk in proportion to the lowering of atherogenic lipoproteins. It also has been consistently shown that, when discordance exists between LDL-C and ApoB/LDL-P, the risk of disease/events is always more closely associated with the lipoprotein parameters, both in drug-naïve and drug-treated patients. The following algorithm would seem to be a prudent approach to these patients:
Step 1: When faced with an increase in LDL-C in a LCKD patient, assess lipoprotein status with ApoB or NMR- derived LDL-P (to my knowledge, no other form of LDL-P has been validated in clinical disease/outcomes trials as superior to LDL-C in predicting CVD risk). Those without increased ApoB/LDL-P can be reassured that their LDL-C increases are benign.
Step 2 (optional for primary prevention patients ONLY): Imaging studies could be helpful to assess CVD status. Coronary artery calcium and cIMT studies are regularly available, and some are using CT angiogram studies. The presence (or absence) of subclinical atherosclerosis could be used to modify one’s approach as to the intensity of treatment of those with elevated increased LDL-C/ApoB/LDL- P. It should be remembered, though, that no imaging study has ever been clinically validated as a way to judge the efficacy of any therapy and no guidelines have recommended follow-up or serial imaging studies as a way to evaluate the efficacy of any therapy.
Step 3: For those with increased ApoB/ LDL-P, the following measures can be taken:
- Decrease saturated fat intake, especially saturated fat known to increase LDL-C the most, (e.g. dairy products and coconut oils). Increase poly- and mono-unsaturated fats without increasing total carbohydrates.
- Increase protein intake as a proportion of daily calories.
- Liberalize carbohydrate intake.
- Re-assess ApoB/LDL-P status. (Some have suggested that the atherogenic particle increases are the result of the anorexic effect of these diets and may normalize after six months.)
Step 4: Treat those with persistently increased non-HDL-C/ApoB/LDL-P levels per the National Lipid Association Recommendations for Patient-Centered Management of Dyslipidemia, usually with statin therapy, to achieve recommended target levels.
Disclosure statement: Dr. Pokrywka has received consultant and speaker honoraria from AstraZeneca, Amarin, Regeneron, and Amgen. He has received speaker honoraria from Kowa Pharmaceuticals America and Health Diagnostic Laboratory Inc.
References are listed on page 31 of the PDF.
Article By:

Assistant Professor, Johns Hopkins University School of Medicine
Baltimore, MD
Diplomate, American Board of Clinical Lipidology