




.jpg)
.png)










When treating our lipid patients it is important to always consider unusual causes of dyslipidemia in our differential diagnosis. Although rare, lysosomal acid lipase deficiency (LAL-D) is often misdiagnosed as an autosomal dominant lipid disorder and/or fatty liver disease.1 LAL-D is caused by genetic mutations that result in a marked decrease or loss in LAL enzyme activity in the lysosomes across multiple body tissues resulting in multi- organ damage and premature death.2,3 With enzyme replacement therapy now available for LAL-D, the outcomes related to this devastating disease are more hopeful.
Lysosomes are membrane-enclosed organelles that contain an arrangement of enzymes capable of breaking down proteins, nucleic acids, carbohydrates, and lipids. Lysosomal Storage Disorders (LSDs) are inherited conditions in which one or more of the enzymes in lysosomes are deficient or not functioning effectively. When this occurs, materials that would normally be hydrolyzed by the lysosome accumulate causing disturbances in normal cell function.2 The Lysosomal acid lipase (LAL) enzyme plays an important role in metabolizing cholesteryl esters (CE) and triglycerides (TG) in the liver, spleen, blood vessel walls, and other organs.2,3 When this enzyme is absent or deficient, the accumulation of CE and TG in the hepatocytes can lead to steatosis, which can progress to cirrhosis.4,5 Lipoprotein cholesterol is primarily in the form of CE and any malfunction or deficiency in this pathway can affect lipid metabolism and the formation of atherosclerosis (Figure 1).6
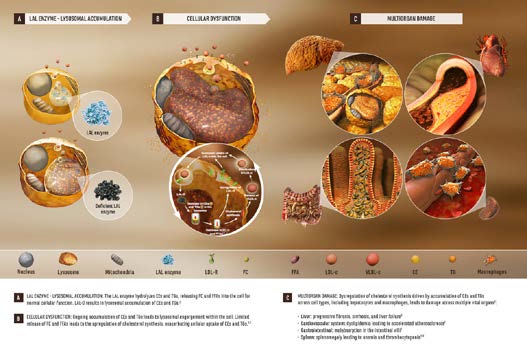
LAL-D is a rare autosomal recessive lysosomal storage disease caused by mutations in the lipase A, lysosomal acid, cholesterol esterase (LIPA) gene.1-5 These gene mutations can manifest in infancy, known as Wolman Disease, and is fatal soon after birth without enzyme replacement. A more common non-fatal expression occurs in children and adults and is known as Cholester yl Ester Storage Disease (CESD).2,3,5 The age-related difference in the progression of the disease is thought to be associated with the nature of the mutations and the degree of residual enzyme activity.2 Dyslipidemia is a common finding in patients with LAL-D and is often linked to the early development of atherosclerosis, cardiovascular disease, and premature mortality.1-5 This condition often goes unrecognized or is misdiagnosed as heterozygous familial hypercholesterolemia (HeFH) due to the similarities in the lipid profile. In addition, the physical characteristics of the disorder are non-specific and can overlap with other diseases.1,2,3
A timely, accurate diagnosis is essential for treatment of LAL-D to lessen the cardiovascular and hepatocellular sequelae of the disease. The clinical evaluation of these patients demonstrates a type IIa or type IIb hyperlipidemia with high total cholesterol, elevated LDL-C, elevated ApoB, and decreased HDL-C levels.2 In addition, elevated levels of serum alanine aminotransferase (ALT) and/or aspartate aminotransferase (AST) are observed with with var ying levels of liver damage.2,3 Hepatomegaly and splenomegaly are seen in nearly all children and adults with LAL-D. A characteristic feature is the presence of markedly hypertrophic Kupffer cells and portal macrophages on liver biopsy.2
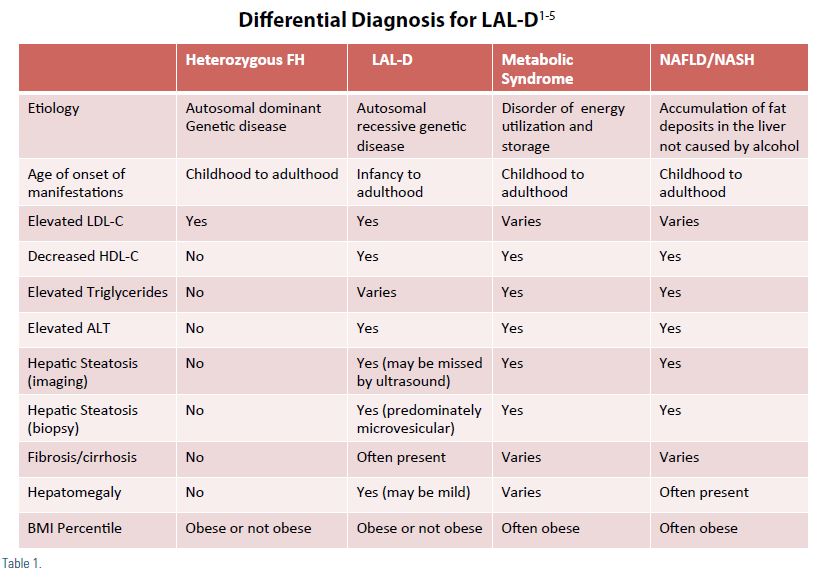
The clinical findings in LAL-D would lead to differential diagnosis including; HeFH, familial defective ApoB, familial combined hypercholesterol (FCH), metabolic syndrome, as well as non- alcoholic fatty liver disease (NAFLD), nonalcoholic steatohepatitis (NASH), and Wilson’s disease.1-5 The astute clinician will get a thorough family history to help differentiate between autosomal dominant disorders (e.g. HeFH, FCH, and polygenic hypercholesterolemia) from autosomal recessive disorders (e.g. LAL-D and sitosterolaemia).2 A complete viral/immunological profile is important to rule out other etiologies such as hepatitis and autoimmune liver disease. A clinical pearl when reviewing the lipid panel of a patient with LAL-D; the total cholesterol and LDL-C are often not as high as seen with HeFH. Also, the HDL-C is usually lower in LAL-D then in HeFH. Another differential clue is that LAL-D patients with dyslipidemia, fatty liver disease, and elevated LFTs are usually not obese as is commonly seen in our metabolic syndrome patients (Table 1).1-5
Screening for LAL-D involves both clinical knowledge and, if indicated, measurement of LAL activity and genetic testing. Z Reiner has recommended a clinical algorithm for deciding upon genetic screening test for LAL-D. Initially, when evaluating a patient with dyslipidemia, it is important to determine the absence or presence of family histor y of hyperlipidemia. The absence of family histor y is more indicative of an autosomal recessive trait such as LAL-D, particularly if the patient has also tested negative for autosomal dominant genetic causes such as HeFH and FCH.2 The next step in the algorithm is to assess the patient utilizing the following criteria:
- ALT > 1.5 X upper limit of normal
- Hepatomegaly present (may be mild)
- HDL-C < 50mg/dL
- BMI < 30 kg/m
- Liver biopsy (if carried out) findings suggestive of microvesicular steatosis
The patient only needs to meet three of the criteria to warrant further genetic testing of LAL activity or mutations in the LIPA gene.
Previous treatment of LAL-D has been limited to supportive therapy to reduce the burden of disease and the use of HMG- CoA reductase inhibitors (statins) alone or in combination with other lipid-lowering therapies. These medications reduce serum cholesterol and TG numbers; however, this lowering effect has not been consistent with improvements in liver function tests or hepatic CE or TG content.4 Several studies have demonstrated that treatment with statins does not change the progression of liver damage in patients with LAL-D.2,4
In December 2015, the Food and Drug Administration approved sebelipase alfa as the first treatment for patients with lysosomal acid lipase (LAL) deficiency. Treatment is provided via intravenous infusion once weekly in patients with rapidly progressive LAL deficiency presenting in the first six months of life, and once ever y other week in all other patients.6 Sebelipase alfa is an innovative enzyme replacement therapy that addresses the underlying cause of lysosomal acid lipase deficiency by reducing substrate accumulation in the lysosomes of cells throughout the body.6 In the multicenter, randomized, placebo- controlled ARISE (Acid Lipase Replacement Investigating Safety and Efficacy) study of children and adults with LAL-D, treatment with sebelipase alfa improved survival in infants with LAL-D and led to significant reductions in ALT and liver fat content, as well as significant improvements in lipid parameters, in children and adults with LAL-D.6
Lysosomal acid lipase deficiency should be included in the differential diagnosis for all patients with increased serum total and LDL cholesterol with a marginally low HDL, elevated AST/ALT and hepatomegaly. Although LAL-D is an ultra-rare disease, affecting two to 25 lives per million depending on the time of onset, increased awareness of this condition and the evidence based enzyme replacement therapy now available is important in reducing the devastating effects of this disease.
Disclosure statement: Dr. Friedrich has no disclosures to report.
References are listed on page 32.
Article By:

DNP Post-Master’s Director
University of South Florida, College of Nursing
Bradenton, FL
Diplomate, American Board of Clinical Lipidology