





.jpg)
.png)










Lipoprotein(a), also referred to as Lp(a), is an unusual plasma lipoprotein that was first described by Berg in 1963.1 The lipoprotein(a) particle consists of a low density lipoprotein (LDL) particle to which a single molecule of apoprotein(a) is covalently bound via a disulfide linkage to apoprotein B-100. The size of the apoprotein(a) moiety varies substantially between individuals because of differences in the number of kringle-4 repeats, as discussed below. Lipoprotein(a) is formed in plasma, possibly on the surface of hepatocytes, primarily from circulating LDL and hepatically secreted apoprotein(a). The distribution of plasma concentrations of lipoprotein(a) in the general population is highly skewed toward zero, with the range varying more than 1000-fold. The median concentration in Caucasians, Asians, and Hispanics is 10 to 20 mg/dL, with levels being 2-3 fold higher among blacks.2
The normal function of lipoprotein(a) is uncertain, since there is no clear deficiency state, most animal species do not produce lipoprotein(a) (it is found only in humans, apes, old world monkeys, and European hedgehogs), and most humans have low concentrations of plasma lipoprotein(a). It is has been proposed that lipoprotein(a) may function to deliver cholesterol to sites of injury and repair in various tissues, but there are other mechanisms for accomplishing this task in the absence of lipoprotein(a). Anticarcinogenic properties have been proposed for lipoprotein(a), and the results of one recent study showed a significant association between prospective cancer risk and low concentrations of lipoprotein(a) in 10,413 participants followed for a median of 12.5 years3, but most studies have shown no association. Lipoprotein(a) is of interest to lipidologists and other health care providers because it is a risk factor for and mediator of thrombosis and accelerated atherogenesis.

Assays for Lipoprotein(a)
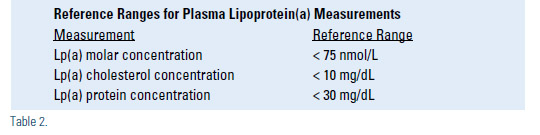
Measurements of lipoprotein(a) cannot be interpreted without an understanding of the diverse variations in laboratory methodology. Measurements of plasma lipoprotein(a) concentrations are performed by several different methods, which has been a significant source of ambiguity and confusion in interpreting published data and diagnostic results provided by various laboratories.4 There has been some success in standardizing the quantitative assays used for measuring lipoprotein(a) concentrations, but variability between laboratories can still produce disparate results.4 In addition, various laboratories provide results in units that are not directly interchangeable. The three most common assay units utilized are nmol/L of lipoprotein(a) particles, mg/dL of lipoprotein(a) protein (usually a measurement of apoprotein(a) by ELISA), and mg/dL of lipoprotein(a) cholesterol. The latter two differ about 3-fold, but the results are easily confused because both are expressed in units of mg/dL, often without designation of measurement of protein or cholesterol. One mg/dL of apoprotein(a) protein is comparable to about 2.4 nmol/L of lipoprotein(a), but the proportion varies from 1.8 for large apoprotein(a) size to 2.9 for small apoprotein(a). Other methods of assessing lipoprotein(a) include determination of the apoprotein(a) genotype and quantification of the number of kringle-4 repeats in the apoprotein(a) molecule. It is estimated that the apoprotein(a) genotype alone accounts for 90% of heterogeneity in plasma concentrations of lipoprotein(a), and the results of family studies provide a similar estimate of heritability of lipoprotein(a) levels. Other causes of elevated concentrations of lipoprotein(a) are shown in Table 1. The molar plasma concentration of lipoprotein(a) is inversely proportional to the number of kringle-4 repeats in the apoprotein(a) molecule, which means that the largest apoprotein(a) molecules are associated with the lowest concentrations of lipoprotein(a) in plasma. Practitioners need to familiarize themselves with the assay used by their laboratory, including the accuracy and reproducibility of the results, so they can correctly interpret the lipoprotein(a) results from their patients. Reference ranges for lipoprotein(a) are shown in Table 2.
Lipoprotein(a) and Cardiovascular Risk
Lipoprotein(a) plays a causative role in atherogenesis and cardiovascular disease (CVD) through several mechanisms related to increased thrombogenesis and lipid deposition in the artery wall.5-8 The risks of coronary artery disease, cerebrovascular disease, and peripheral vascular disease are all increased in the setting of high levels of lipoprotein(a). Up to 20% of individuals with early onset CVD have high levels of lipoprotein(a) > the 95th percentile, which demonstrates that elevated lipoprotein(a) is fairly common in this patient population.
The apoprotein(a) molecule is a homologue of the fibrinolytic proenzyme, plasminogen, the precursor of plasmin. The presence of high levels of apoprotein(a) can interfere with plasminogen activation and thereby contribute to thrombosis by decreasing fibrinolysis and enhancing clot stabilization. Lipoprotein(a) may also interfere with the function of tissue factor pathway inhibitor, which increases thrombogenesis. Accordingly, very high concentrations of lipoprotein(a) can be associated with spontaneous arterial thromboses, and possibly venous thromboses, but a recent Mendelian randomization study of lipoprotein(a) genotype and plasma concentrations in 41,231 individuals did not demonstrate a relationship between lipoprotein(a) and venous thrombosis except when lipoprotein(a) levels were greater than the 95th percentile.9
Lipoprotein(a) also plays an important role in atherogenesis, particularly in the presence of elevated concentrations of LDL or remnant lipoproteins.10,11 The lipoprotein(a) particle appears to be more readily retained in the artery wall and it accumulates at sites of arterial injury or inflammation. In addition to its atherogenic cargo of cholesterol, lipoprotein(a) is also a carrier of pro-atherogenic oxidized phospholipids and lipoprotein-associated phospholipase A2 (Lp-PLA2; also known as PAF acetylhydrolase). Several lines of evidence suggest that the risk of CVD appears to be related to a synergistic relationship between lipoprotein(a) and LDL, as reflected by the attenuation of risk in individuals with high lipoprotein(a) but low LDL-C10,11, the enhancement of risk in subjects with heterozygous familial hypercholesterolemia and high lipoprotein(a)12, and the suppression of risk of CVD events by aggressive LDL-C lowering in patients with pre-existing CVD and high lipoprotein(a) concentrations.13
Lipoprotein(a), Aortic Valve Calcification and Aortic Stenosis
The very interesting results of a recent study have suggested that lipoprotein(a) also contributes to aortic valve calcification and incidence of aortic stenosis. Genomewide associations with the presence of aortic valve calcification were assessed in 6942 subjects in 3 cohorts, which led to the identification of a single nucleotide polymorphism in the lipoprotein(a) (LPA) locus (rs10455872) associated with an odds ratio of 2.05 (P=9.0 x 10–10) for aortic valve calcification.19 Lipoprotein(a) levels predicted by the LPA genotype also were associated with aortic valve calcification. In a prospective analysis, the LPA genotype also was associated with the incidence of aortic stenosis with a hazard ratio of 1.54 (95% CI 1.05 to 2.27).
Screening for Lipoprotein(a) Elevation
Screening for lipoprotein(a) elevation is indicated in patients with moderate to high CVD risk because it is helpful for CVD risk stratification and helps guide the aggressiveness of treatment of dyslipidemia. Identification of an individual with high lipoprotein(a) also is a marker of genetically mediated CVD risk, which provides the opportunity for detection of first degree relatives who unknowingly may also have increased CVD risk. Screening of seemingly low-risk patients also needs to be considered because the advent of the statin era 25 years ago has substantially reduced the sensitivity of the family history for detection of familial CVD risk. Since an entire generation of patients have markedly reduced their CVD risk as a consequence of effective LDL-lowering by statins, the offspring of these patients (and their health care providers) can no longer assume that a negative family history of CVD implies low CVD risk. The implication of this is that patients who report having no family history of CVD may actually have increased CVD risk related to lipoprotein(a) elevation or other genetically mediated CVD risk factors. The European Atherosclerosis Society Consensus Panel recently advocated screening all individuals with the following conditions: (I) premature CVD, (II) familial hypercholesterolemia, (III) a family history of premature CVD and/or elevated Lp(a), (IV) recurrent CVD despite statin treatment, (V) > 3% 10-year risk of fatal CVD according to the European guidelines, and (VI) >10% 10-year risk of fatal and/or non-fatal CHD according to the US guidelines.8 A family history of hypercholesterolemia could also be considered as an alternative criterion for item (III) because of the reasons described above. The National Lipid Association also convened a panel of clinical experts who issued recommendations regarding the clinical use of various biomarkers in 2011, which included recommendations for lipoprotein(a) that mirrored the recommendations from the European Atherosclerosis Society Consensus Panel.14

Treatment
There is no direct proof that lowering lipoprotein(a) reduces CVD risk because the studies have not been done. In the meantime, it is reasonable to try to reduce high levels of lipoprotein(a) in selected patients. Niacin is the primary pharmacologic treatment for elevated lipoprotein(a) because it has the greatest lipoprotein(a)-lowering efficacy and it has been shown to reduce CVD events in several patient populations.15 Unfortunately, the efficacy of this intervention is limited to a 20-40% dosedependent reduction, which is insufficient to achieve acceptable levels in patients with very high levels. There are reports that statins minimally lower plasma lipoprotein(a) concentrations, but statins are generally ineffective for lipoprotein(a) lowering except in patients with familial hypercholesterolemia, who may achieve modest lipoprotein(a) lowering for unclear reasons (lipoprotein(a) is not cleared by the LDL receptor). A possible mechanism is decreased production of lipoprotein(a) due to a reduced pool of LDL in plasma.
LDL apheresis can acutely lower lipoprotein(a) levels by 50-80% during a 2-3 hour procedure, but the invasiveness of the procedure, high cost, and fairly limited availability are limiting factors. Individuals with very high lipoprotein(a) concentrations and progressive CVD despite aggressive medical therapy may be candidates for initiation of treatment with LDL apheresis. We are currently treating two individuals with LDL apheresis for this indication, one of whom had severely elevated lipoprotein(a) concentrations and developed rapidly progressive internal carotid artery atherosclerotic occlusions necessitating bilateral revascularizations in her early 50s, despite aggressive combination treatment with a statin plus niacin. In an uncontrolled observational study of 120 patients with CVD and lipoprotein(a) levels greater than the 95th percentile, treatment with LDL apheresis was associated with a reduction in CVD events (MACE rate per patient 1.056 vs. 0.144; P<0.0001).16 The results of a more recent randomized trial of LDL apheresis in 32 patients with lipoprotein(a) > 50 mg/dL and LDL cholesterol < 2.5 mmol/L demonstrated increased regression or stabilization of angiographic coronary atherosclerosis (70% vs 43%, p=0.02) compared to usual care.17
Treatment with estrogen replacement or estrogen analogues in postmenopausal women is associated with a modest reduction in the plasma concentration of lipoprotein(a), but the predictive association between lipoprotein(a) and cardiovascular risk is attenuated in women taking hormone replacement therapy.18 Among postmenopausal women with the highest quintile of lipoprotein(a), however, those women taking hormone replacement appeared to have a lower risk of cardiovascular events compared to those not taking estrogen, particularly among women with high LDL cholesterol concentrations above the median.18 The relationship between estrogen replacement and CVD risk in the general population continues to be controversial, however. Treatment of hypothyroidism, if detected, and correction of renal insufficiency and proteinuria, if possible, may also have beneficial effects on lipoprotein(a) levels. Anabolic steroids such as stanozolol and danazol may lower lipoprotein(a) levels up to 50% in women, but these agents are not recommended for general use because of adverse side-effects. It is possible that aspirin, L-carnitine, ascorbic acid combined with L-lysine, calcium antagonists, angiotensin converting enzyme inhibitors, and androgens may lower lipoprotein(a) by < 10%8, but these agents are not indicated as primary treatment for elevated lipoprotein(a). Experimental medications are under development that may lower lipoprotein(a) concentrations by more than 20-25%, such as lomitapide (microsomal transfer protein inhibitor), mipomersen (apoprotein B antisense oligonucleotide), anti-PCSK9 agents, and thyroid hormone analogues. Lomitapide and mipomersen were recently FDA approved for restricted treatment of homozygous familial hypercholesterolemia, but they are still considered experimental for lipoprotein(a) lowering and treatment of other patient populations.
Since it is typically difficult to normalize plasma levels of lipoprotein(a), an alternative strategy is to aggressively lower levels of LDL and remnant lipoproteins in patients with high lipoprotein(a). The efficacy of this strategy is not proven, but it is supported by the findings from prospective observational and intervention studies that suggest that the risk of CVD events attributable to lipoprotein(a) may be abrogated when the LDL cholesterol concentration is < 70-80 mg/dL.10,11,13
Summary and Conclusions
Lipoprotein(a) elevation is an important risk factor for CVD, including coronary artery disease, cerebrovascular disease, and peripheral vascular disease, particularly among individuals with the highest levels of lipoprotein(a) in combination with elevated LDL cholesterol or particle numbers. Levels of plasma lipoprotein(a) are rarely quantified outside of lipid disorder clinics, so the majority of patients with high levels are undiagnosed. About 90% of the inter-individual heterogeneity in levels of lipoprotein(a) is genetically mediated, so the disorder is highly heritable, necessitating screening of first degree relatives of affected individuals. The methods for quantifying lipoprotein(a) are not well standardized, so practitioners need to understand how the testing is performed and what is actually measured in their laboratory. All moderate- and highrisk patients should have a lipoprotein(a) determination, and some seemingly low risk individuals may warrant evaluation. Niacin is the most efficacious treatment for lowering lipoprotein(a), aside from LDL apheresis. Aggressive LDL lowering is an alternative strategy for managing patients with elevated lipoprotein(a), since the atherogenicity of lipoprotein(a) appears to be attenuated when the LDL-C concentration is low. Clinical trials are needed to demonstrate the best approach to managing patients with high lipoprotein(a), but in the meantime we need to utilize the available strategies in the context of our current understanding of lipoprotein(a) and CVD risk.
Disclosure statement: Dr. Duell has received honoraria from Aegerion, Genzyme, Merck & Co., and Vivus. Dr. Duell has received grants from Bristol-Myers Squibb, Cerenis, Pfizer Inc., and Genzyme.
Article By:

Director, Lipid Disorders Clinic and Lipid-Atherosclerosis Laboratory
Oregon Health and Science University
Portland, OR